Metabolic effects of Redostim
Synthesis of GSH and H2S
Redostim’s micronutrients support both GSH and H2S synthesis in response to the normal physiological stimuli that induce them.
GSH synthesis is activated when the cellular environment becomes oxidizing (redox-regulated). GSH is a very simple water-soluble molecule capable of bringing reducing power (antioxidant) wherever it is needed. Basically, it recharges many of the other cellular antioxidants, both fat-soluble (e.g. Vit. E) and water-soluble (e.g. Vit. C) and is the main physiological antioxidant effector. GSH cannot be “eaten”, it must be produced in the cell as needed, but we can take the substances that promote its synthesis. The effect of Redostim on GSH release can be monitored by contacting common laboratories that can measure both plasma and erythrocytic GSH.
Hydrogen sulphide (H2S) is released by the same enzymes that synthesize GSH when they work according to their alternative mode. This happens when cells undergo various types of stresses including oxy-redox (in both directions), thermal, reticuloendothelial (protein misfolding), and others. The H2S released by a cell spreads (it is a gas) to nearby ones, influencing their activity by epigenetic modifications (activation / repression of genes) and by modifications of the activity of functional proteins. Given the nature of its release, the effects of Redostim on H2S release cannot be measured with the circulating level of the substance which is erratic, difficult to measure accurately and of little clinical significance, and in fact the common laboratories do not offer this test. Instead, it is possible to measure the concentration in the blood of the substances produced during its release. This can be done only through mass spectrometry methods and makes sense only in particular situations (e.g. subjects with high cardiovascular risk). Interested parties can contact us by writing to testing@parthenogen.eu

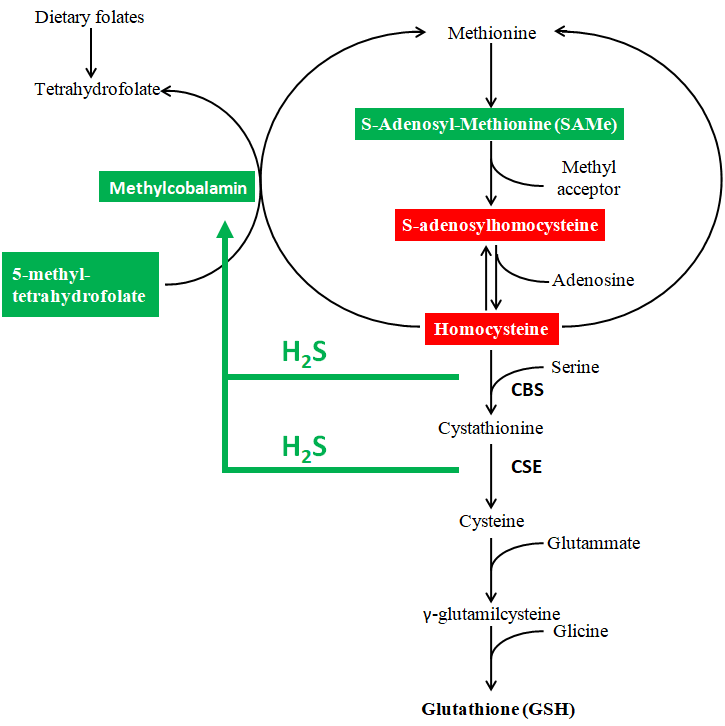
Carbon cycle, methylations and epigenetics
H2S is a powerful inducer of the carbon cycle (or methionine / folate cycle). The best-known reason for this effect is the induction of the activation of vitamin B12 to methylcobalamin, i.e. the form capable of passing the methyl group to homocysteine forming methionine. In turn, methylcobalamin will feed the methylations that are responsible for fundamental chemical syntheses (DNA, RNA, creatine, choline, CoQ, carnitine) and for DNA methylation in the context of epigenetic regulations.
The intake of Redostim induces a strong increase in methylcobalamin, an increase in S-adenosyl methionine (activated methyl donor) metifolate (active form for the remethylation of homocysteine) with reduction of S-adenosyl homocysteine and homocysteine (both inhibitors of methylations).
Induction of nitric oxide (NO)
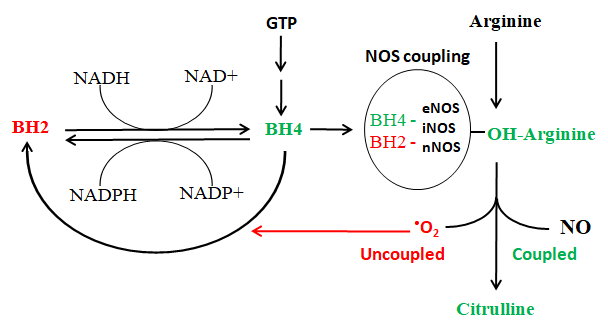
Nitric oxide is, like H2S, a gas and a mediator of cellular homeostasis. NO acts in concert with H2S in regulating endothelial function. Deficiency of NO or NO activity is known to be implicated in the development of cardiovascular disease and erectile dysfunction.
The synthesis of NO is complex and is performed by 3 enzyme isoforms of the nitric oxide synthase (NOS), respectively endothelial (eNOS), neuronal (nNOS) and inducible (iNOS). NOS metabolizes the amino acid arginine, transforming it first into hydroxy-arginine and then into citrulline. This step requires the presence of a cofactor, the tetrahydro-pteridine (BH4) which acts as an intermediate electron acceptor. In the presence of BH4 the NOS produce citrulline and NO. If, BH4 is present in its oxidized form, BH2, the reaction produces instead of NO the powerful radical superoxide anion (•O2). The superoxide anion goes to oxidize BH4 to BH2 creating a vicious circle that increasingly reduces the production of nitric oxide to the benefit of superoxide. BH2 can be reduced back to BH4 by the same enzyme that activates folates, DHFR.
The synthesis of nitric oxide does not increase by administering arginine, indeed an excess of arginine is used to produce uncoupled reactions and generates superoxide and a degradation product of arginine (ADMA) with strong cardiovascular toxicity. The main regulator of NO synthesis is BH4 and the ratio BH4: BH2. Also for nitric oxide the best monitoring system is the study in mass spectrometry of the concentration of the products of its synthesis, in this case arginine, citrulline, BH4 and BH2.
The micronutrients of Redostim induce a strong increase in BH4, a reduction in BH2 and an increase in the ratio BH4: BH2, and this strongly activates the release of NO. This is confirmed by the increase in citrulline (accumulation of the reaction product) without changes in arginine (consumption in the reaction).
Homocysteine re-cycling
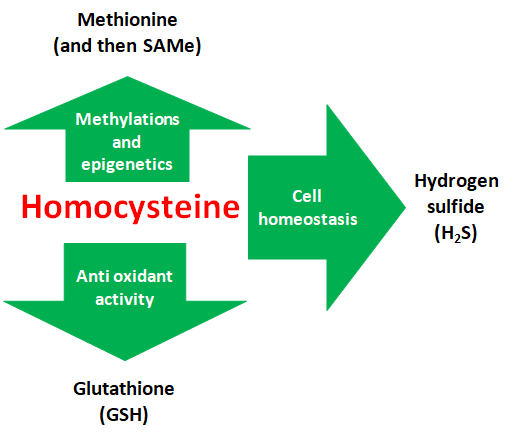
Homocysteine is a non-proteogenic amino acid that is formed as a residual product in methylation reactions (addition of carbonaceous unit to a molecule). It is a known cardiovascular risk factor and has been linked to almost all degenerative and aging diseases. It is in fact a powerful methylation inhibitor (epigenetic interferent) and a powerful pro-oxidant. It is massively produced in all cells and equally massively removed from the metabolism. The fasting plasma homocysteine dosage is offered by all the common laboratories which, however, rarely perform the dosage on urine, which would be more informative: many subjects have only post-prandial hyperhomocysteinemia which does not result testing morning fasting homocysteine. The presence of fasting homocysteinemia above normal limits always indicates a metabolic imbalance. Very high hyperhomocysteinemia is always linked to genetic defects of the enzymes that dispose of it, the disease called homocystinuria is generally linked to mutations of the CBS enzyme. Mild and moderate hyperhomocysteinemias can be result from incorrect eating habits and can be corrected with the diet, but these subjects often also carry a predisposing genetic substrate.
Homocysteine is disposed of in three different ways: i) it is re-methylated to methionine (methionine / folate / betaine cycle); ii) it is used to synthesize cysteine, this in turn is incorporated into proteins or used to synthesize GSH (antioxidant defenses); iii) two homocysteine molecules or a homocysteine and a cysteine can bind and in doing so release H2S. The micronutrients of Redostim supply all three of these homocysteine removal pathways and favor their “positive” reuse to feed methylations, antioxidant activity and H2S.
Homocysteine is disposed of in three different ways: i) it is re-methylated to methionine (methionine / folate / betaine cycle); ii) it is used to synthesize cysteine, this in turn is incorporated into proteins or used to synthesize GSH (antioxidant defenses); iii) two homocysteine molecules or a homocysteine and a cysteine can bind and in doing so release H2S. The micronutrients of Redostim supply all three of these homocysteine removal pathways and favor their “positive” reuse to feed methylations, antioxidant activity and H2S.
Regulation of oxy-redox equilibrium
The classic interpretation of oxidative stress, the one based on the idea that we are bombed by the oxidative load and strive strenuously to defend ourselves by taking on various types of plant antioxidants, is a mere cultural construct. It is great for animating health-oriented talk shows but has no correspondence with contemporary understanding of these phenomena.
The reality is that the real food source of antioxidant activity are the cysteines (SH groups) contained in food proteins: vegetable proteins are more enriched by them, there is no difference between meat and fish proteins (false myth). Most plant antioxidants are not such in humans (they are not physiological antioxidants) and would be interferents. In reality they are little or nothing absorbed and, even in a vegetarian diet, they do not create interference. But beware of supplements.
The physiological antioxidant system is essentially based on three mediators: the first is GSH, which incorporates the reducing power of cysteines and which in turn “recharges” the two oxidation-reduction coenzymes NADH and NADPH. These donate (or accept, when in their oxidized form NAD+ and NADP+) electrons in chemical reactions. The reduction of GSH / NADH / NADPH compared to the needs configures an oxidative stress, their excessive increase configures a reducing stress. Both phenomena are common and lead to the same consequences: the formation of reactive radicals that can “rip off” electrons from oxygen and nitrogen to form free radicals. In essence, the production of reactive oxygen species (ROS) can be a consequence of both oxidative stress and reductive stress. Similarly, the damage these cause to lipids and proteins DNA are the same, regardless of whether oxidative or reductive stress originated them.
When we are short in reducing activity, the synthesis of cysteine starting from homocysteine and the synthesis of GSH starting from cysteine is activated. The GSH produced will then supply NADH and NADPH and restore their levels. The way we react to excess reducing activity is less clear, but the most advanced hypothesis is that this occurs with the release of H2S. In fact, even if H2S has a reducing power similar to that of GSH, it does not seem to behave as a cytoplasmic reducing agent and is unable to pass the reducing power to NAD+ and NADP+. The reducing equivalents transformed into H2S can enter the mitochondria to be oxidized on the respiratory chain to produce ATP, exactly as happens in the organisms that populate the ocean hydrothermal vents. In this way the excess SH groups are used to produce energy and definitively eliminated from the system (correction of the reductive imbalance) by passing into urine as sulphates (SO4). Furthermore, H2S stimulates the release of NO and CO, which are oxidants and further counterbalance the reductive stress.
Redostim’s micronutrients are certainly a support for antioxidant activity because they induce GSH, which is the main physiological antioxidant, and H2S, which is itself a powerful reducing agent. Indeed, according to our data, Redostim is the most efficient among known antioxidant supports. However, the same micronutrients are also capable of promoting the disposal of H2S in the mitochondria for the production of energy / ATP. The administration of these micronutrients is associated with an increase in 3-mercaptopyruvate, a product of the transamination of cysteine that enters the mitochondria and here releases H2S, in parallel with an increase in sulphates, a product of the oxidation of H2S in the mitochondria, and its use for the ATP synthesis. In essence, Redostim’s micronutrients provide cells with the tools to balance both oxidative and reductive stresses. It will be up the single cells, depending on their state, to decide whether to use them in one direction or the other by obeying to the physiological homeostatic signals.

“Buffer” effect on oxy-redox balance – LThe effect of Redostim’s micronutrients on cellular redox balance changes as a function of what the cell needs. If an oxidative imbalance is present, the homeostatic signals will direct the micronutrients mainly towards the synthesis of GSH which will supply the whole cellular antioxidant system. If, on the other hand, there is an imbalance in the reductive sense, they will be mainly directed towards the release of H2S, which will be used by the mitochondria to produce ATP thus eliminating the excess of reducing activity. Note that the effect can be alternately in opposite directions in the same subject at different times and in the same subject and at the same time in different cells / tissues / organs according to their specific needs. In essence, a sort of metabolic buffer system is created which maintains optimal conditions.

“Buffering effect” in mitochondria – The effect of Redostim micronutrients on the oxy-redox balance is the same as described for the cell in general. Mmicronutrients will mainly boost the synthesis of GSH or of H2S according to the needs thus contributing to the resolution of a possible mitochondrial stress.
Correction of mitochondrial dysfunction
The cells produce their energy in the mitochondria, organelles of bacterial origin that contain their DNA and take care of transforming the metabolic energy contained in fats and sugars into ATP, which is the “fuel” of the cells. When mitochondria fail to produce the amount of ATP that the cell requires, we speak of mitochondrial dysfunction. Under these conditions many cellular processes slow down or stop generating pathological consequences. For example, the lack of energy to synthesize DNA can reduce the rate of cell proliferation. Furthermore, a lack of energy during a cell division can cause the arrest of the migration of some chromosomes with the onset of non-disjunction errors that lead to the appearance of cells with an altered number of chromosomes. When this happens in gametogenesis, the entire new organism produced by the gamete will have a different number of chromosomes, in this case we speak of aneuploidy.
The best known reason for the appearance of mitochondrial dysfunction is the accumulation of ROS in the mitochondria. ROS are physiologically produced during the release of ATP, but they do not pass the membrane and become trapped there. It will be up to the mitochondrial GSH to neutralize the accumulated ROS. If mitochondrial GSH is not enough, the mitochondrion slows down and goes into dysfunction.
However, the mitochondrion may also malfunction due to an excess of energy production and reducing stress. When digesting food (Krebs cycle) it transforms chemical energy into reducing power, NADH, and it is this reducing power that oxidizes oxygen to water in exchange for ATP. If the mitochondrion produces too much NADH, station II of the respiratory chain, CoQ10, is in reduced form and is no longer able to pass the electrons to station III. In these conditions it passes reducing equivalents directly to oxygen generating superoxide anion (•O2). In this case, it will be necessary to decrease the concentration of NADH by converting its reducing equivalents to H2S, in this way the NADH is decreased and the ATP release increases thanks to the contribution of H2S.